常见细胞污染类型如何辨别及预防解决方法
常见细胞污染类型如何辨别及预防解决方法:细胞培养中常见的生物污染类型有7种,分别是细菌污染,支原体污染,原虫污染,黑胶虫污染,真菌污染,病毒污染以及非细胞污染,真菌污染来源,一般是来自实验服,并且具有气候性,多雨······

发布时间:2025-12-06 09:00:04 细胞资源库平台 访问量:207
N6-甲基腺苷(m6A)是真核细胞mRNA上最普遍的内部修饰,在神经系统中扮演着重要角色。已有研究表明,m6A在皮层神经发生、小脑发育、成年神经发生、记忆形成和巩固以及轴突再生等多种生物学过程中起重要作用。特别是在胚胎皮层神经发生过程中,m6A信号调节神经干细胞分化的时间进程,但不影响神经元命运的决定。然而,关于m6A信号在不同脑区神经发生中是否发挥相似或不同作用的问题尚未得到充分理解。
下丘脑是大脑中的进化保守区域,作为能量摄入、消耗和脂肪储存的中央调节器。在下丘脑中,多个核团由不同的神经元集合编排各种功能。其中弓状核(ARC)被认为是调节摄食行为的关键枢纽,其中POMC表达的厌食神经元和NPY/AgRP表达的促食神经元在调节摄食行为和能量平衡方面发挥着重要但相反的作用。下丘脑功能依赖于精确协调的发育过程,而下丘脑发育缺陷已与包括肥胖和糖尿病在内的各种疾病相关。值得注意的是,下丘脑是小鼠和人类大脑中m6A水平最丰富的脑区之一。
一些研究表明,m6A修饰功能障碍可能导致包括肥胖、2型糖尿病和代谢综合征在内的各种代谢疾病。人类基因组关联研究(GWAS)发现,FTO位点的内含子单核苷酸多态性(SNPs)与肥胖个体的体重指数强相关。然而,后续研究表明,这些SNPs不影响FTO表达,而是可能调节两个附近基因IRX3和RPGRIP1L的表达。另一方面,FTO在小鼠中的全局过表达导致体重和脂肪增加以及食物摄入增加。考虑到人类遗传学和小鼠研究的相互矛盾证据,m6A信号是否直接参与调节脂肪和体重以及食物摄入仍不清楚,这需要适当的人类细胞模型来解决。
近期,发布在Cell Stem Cell期刊,题为m6A deficiency impairs hypothalamic neurogenesis of feeding-related neurons in mice and human organoids and leads to adult obesity in mice的研究揭示了m6A信号在小鼠和人类类器官弓状核神经发生中的保守作用,并阐明了食物摄入和能量稳态的外转录组调控的发育基础。
1) 研究团队首先构建了下丘脑Nkx2.1-Cre::Mettl14f/f(M14NKO)条件性敲除小鼠模型,探索m6A在发育中下丘脑中的功能。结果显示,与对照组相比,M14NKO小鼠在早期出生后阶段体重增加加速,成年后表现出明显肥胖。磁共振成像(MRI)分析表明,M14NKO小鼠的体重增加主要归因于脂肪增加,脂肪量是对照小鼠的五倍,而瘦体重相似或略有减少。此外,M14NKO小鼠的腹股沟白色脂肪组织、性腺白色脂肪组织和棕色脂肪组织体积更大,其脂肪细胞表现出更明显的肥大。葡萄糖耐量试验(GTT)和胰岛素耐量试验(ITT)显示M14NKO小鼠出现高血糖和胰岛素抵抗。
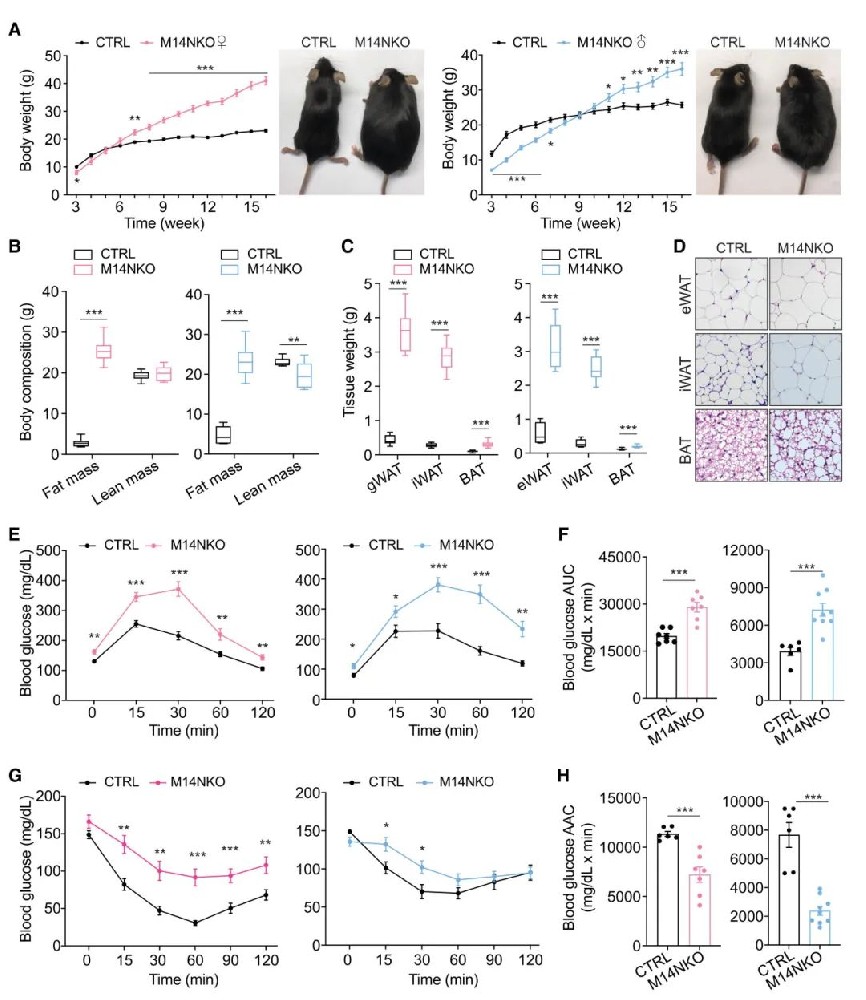
图1 胚胎下丘脑Mettl14缺失导致小鼠成年肥胖和全身葡萄糖稳态功能障碍
2) 代谢分析发现,M14NKO小鼠食物摄入量增加,能量消耗与体重呈正相关,但按体重标准化后,M14NKO小鼠能量消耗减少。氧气消耗(VO2)和二氧化碳产生(VCO2)在M14NKO小鼠中也按体重标准化后降低。雌性M14NKO小鼠呼吸交换率(RER)略有降低,而雄性M14NKO小鼠无变化。此外,成年M14NKO小鼠的运动活动显著减少。这表明胚胎下丘脑Mettl14缺失导致成年小鼠能量平衡受损。
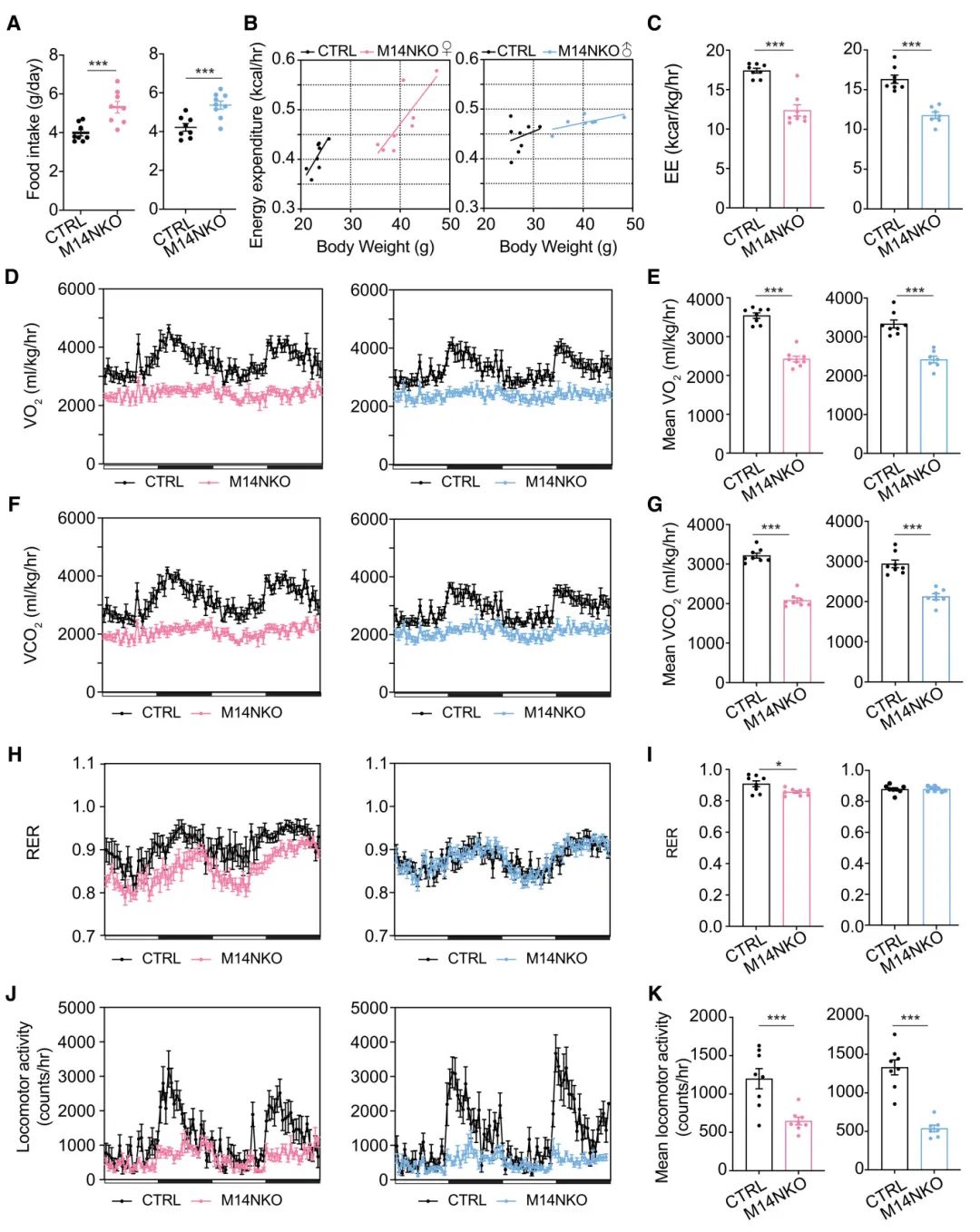
图2 胚胎下丘脑Mettl14消融导致成年小鼠能量平衡受损
3) 为探究M14NKO小鼠肥胖的细胞机制,研究者对ARC区域进行了单细胞RNA测序。分析显示,M14NKO小鼠中ARC神经元的Pomc/Tbx3、Trh/Otp、Arx/Dlx1、Cbln2/Nts、Th/Dlx1、Agrp/Npy/Otp和Avp/Sst集群神经元的比例减少,而Ssr2/Tbx3集群神经元比例增加。Pomc、Agrp、Npy和Otp在相关集群中的表达水平在M14KO小鼠中降低。免疫组织学分析证实,M14NKO小鼠ARC区域的POMC+和OTP+神经元数量减少,但NeuN+神经元总数略有增加。这表明胚胎下丘脑Mettl14缺失导致特定神经元亚型数量选择性减少,而非所有神经元的普遍减少。
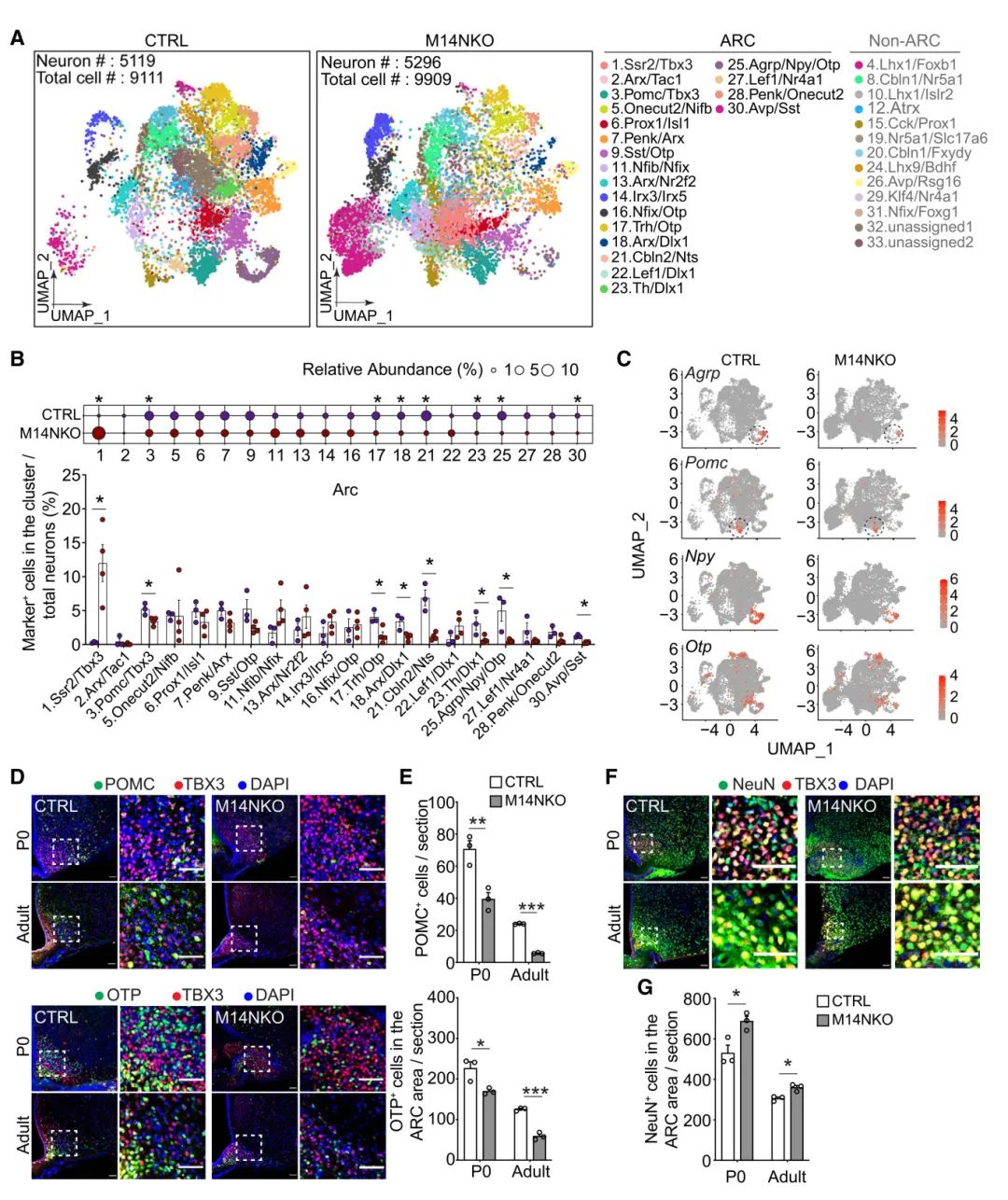
图3 胚胎下丘脑Mettl14缺失导致P0和成年小鼠弓状核中喂养相关神经元数量减少
4) 通过BrdU出生日期标记实验,研究者发现在E12.5注射BrdU时,M14NKO小鼠ARC区域的NeuN+BrdU+神经元数量减少,而在E15.5注射时增加,表明M14NKO小鼠神经发生延迟。重要的是,当在E12.5注射BrdU时,M14NKO小鼠ARC区域的POMC+BrdU+神经元显著减少。此外,在E15.5,M14NKO小鼠ARC前体区域的Ki67+或PH3+细胞数量增加,进一步证实神经发生延迟。这些结果支持Mettl14缺失导致下丘脑神经发生延迟和摄食相关神经元生成缺陷,从而导致成年肥胖。
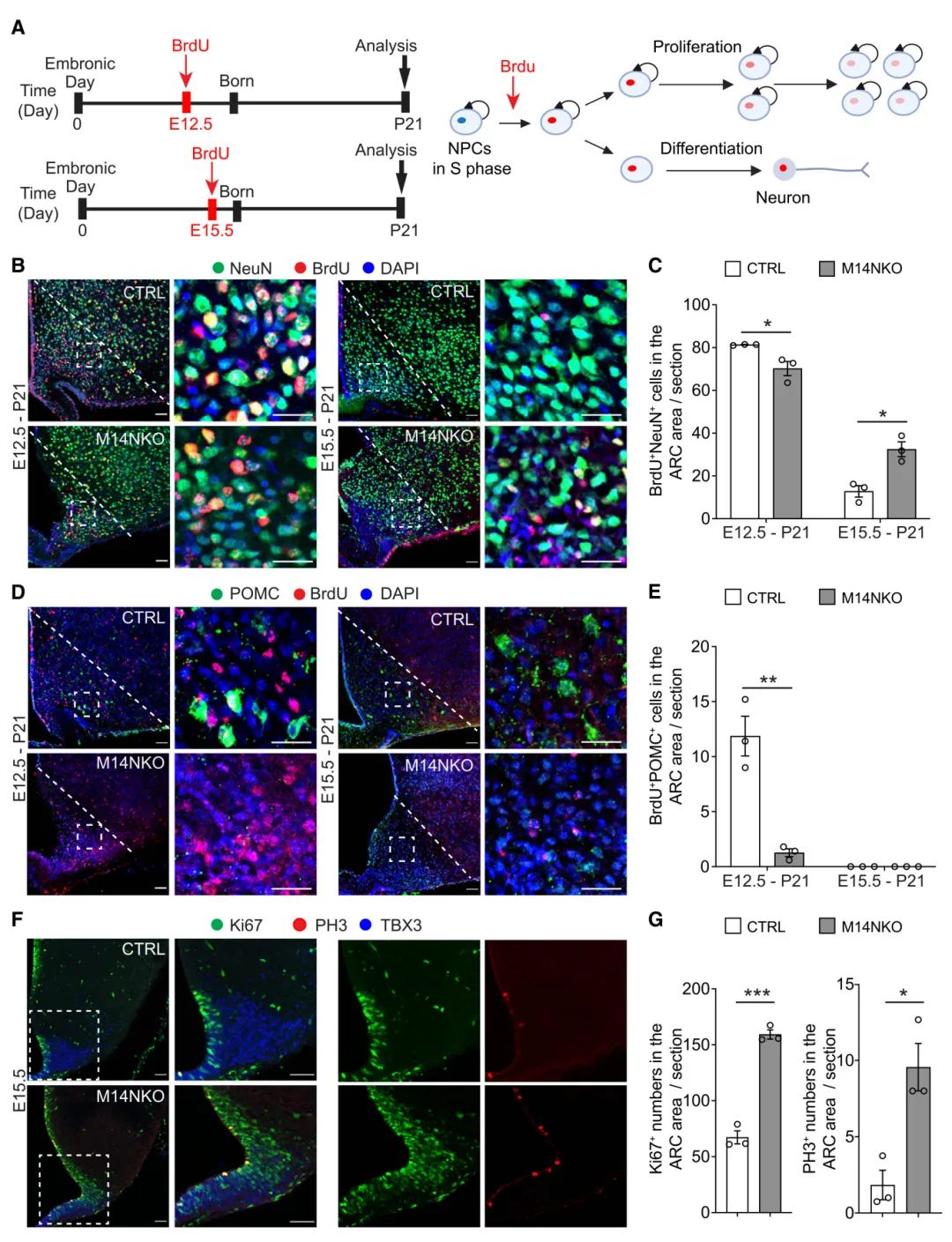
图4 胚胎下丘脑Mettl14缺失会损害了小鼠POMC+神经元的生成
5) 研究小组还分析了Mettl14调节下丘脑神经发生的分子机制。通过整合RNA-seq和m6A-seq数据,他们发现在Mettl14缺失的下丘脑神经前体细胞中,许多与神经系统发育和细胞分化相关的m6A修饰转录本表达下降,包括Wnt信号通路的几个组分(Wnt5a、Wnt5b、Wnt7a和Wnt7b)。这表明m6A可能通过调控关键发育基因的表达来影响下丘脑神经发生。
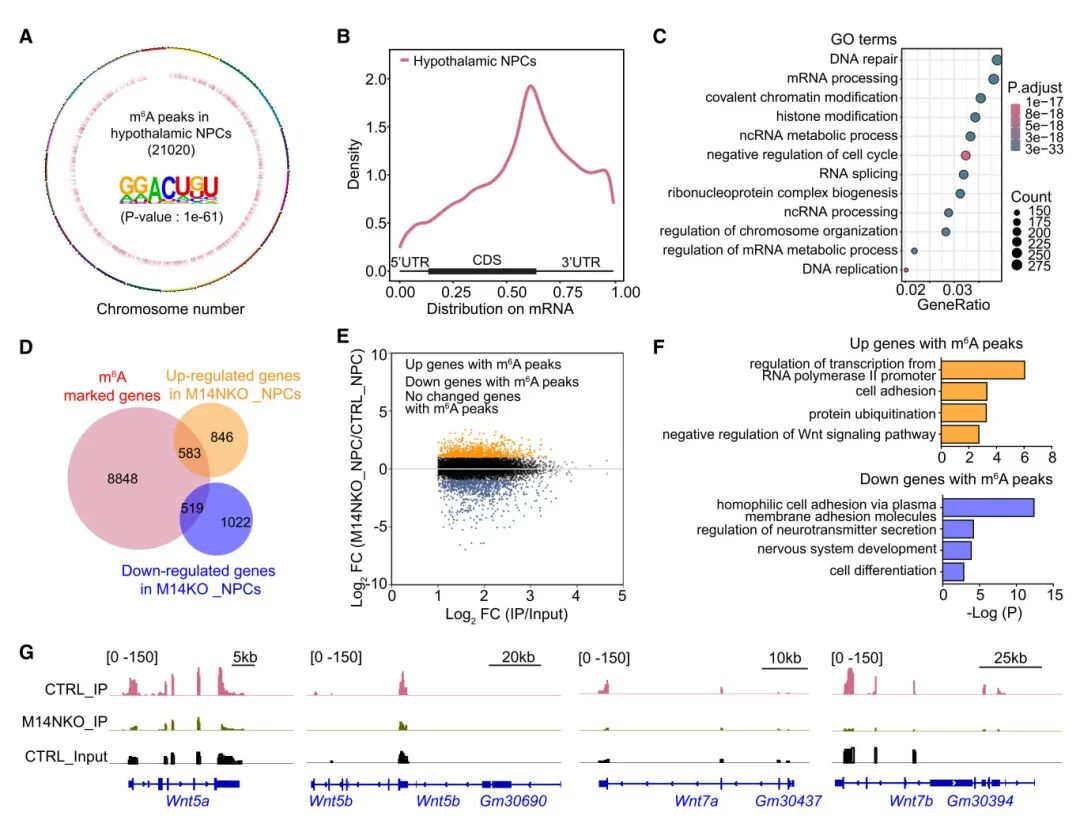
图5 小鼠胚胎下丘脑NPCs中一些m6A标记转录本的表达改变
6) 为确认m6A信号在调节摄食相关神经元生成和体重或脂肪量中的直接作用,研究者还进行了m6A信号通路其他组分的条件性敲除实验。结果显示,敲除另一个m6A写手酶Mettl3(NKX2.1-Cre::Mettl3f/f,M3NKO)的小鼠表现出与M14NKO小鼠相似的成年肥胖和ARC区域POMC+神经元减少的表型。而敲除m6A阅读器Ythdc1(NKX2.1-Cre::Ythdc1f/f,DC1NKO)的小鼠虽然体重与对照组相似,但脂肪量增加,伴随脂肪细胞肥大,食物摄入量增加,以及ARC区域POMC+和OTP+神经元数量减少。这进一步证实了m6A信号通路在下丘脑神经发生和成年肥胖中的关键作用。
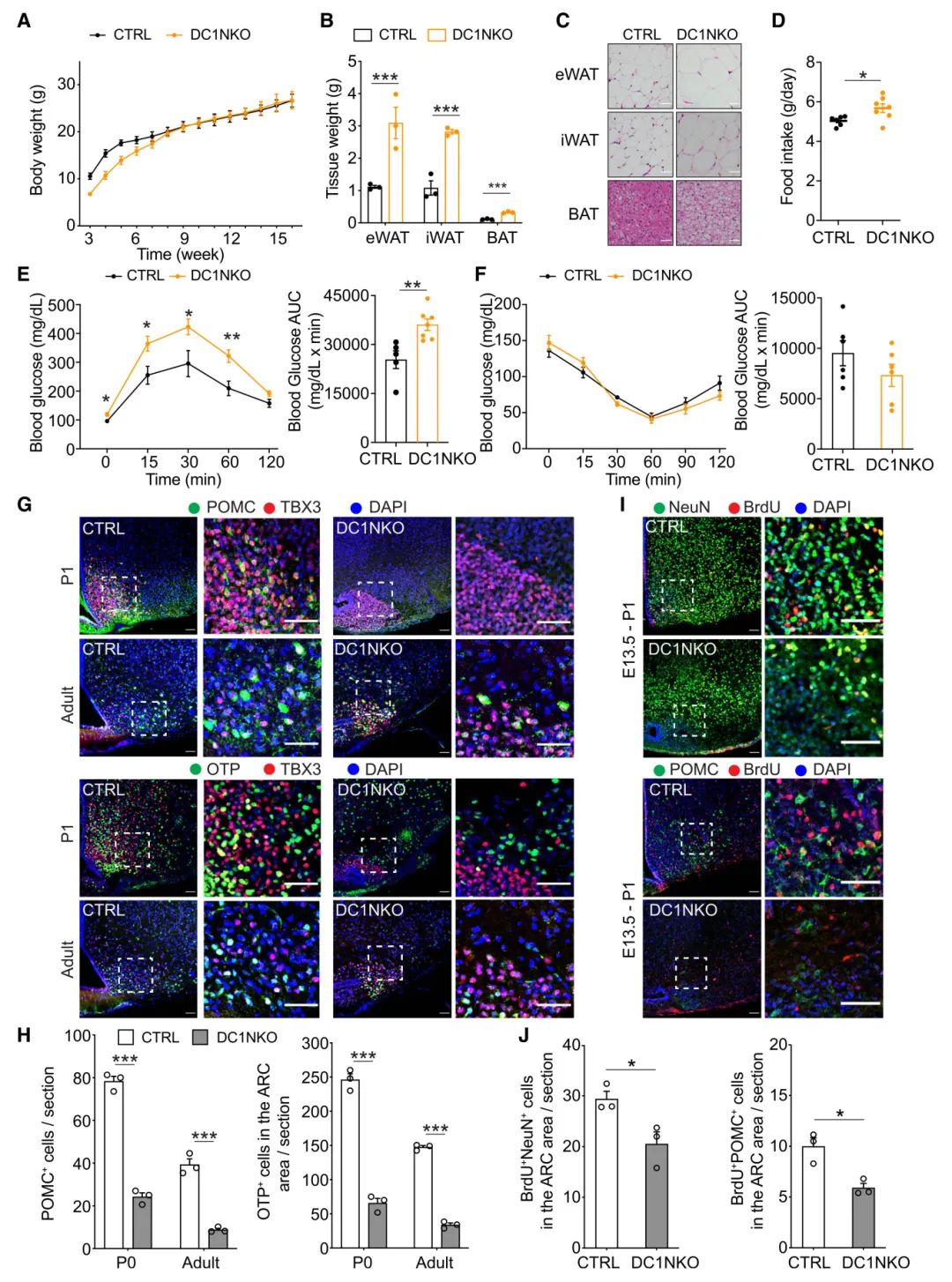
图6 胚胎下丘脑Ythdc1的缺失会损害成年小鼠的喂养相关神经元的生成,并增加脂肪的积累
7) 最后,为探究m6A信号在调节摄食相关神经元生成方面的保守作用是否延伸到人类,研究者使用改进的人类iPSC衍生的脑区特异性ARC类器官(ARCO)模型。他们通过m6A测序确定了人类和小鼠下丘脑神经前体细胞中共同的m6A修饰转录本,包括与染色质重塑、细胞周期调节、DNA修复、RNA剪接和Wnt信号通路相关的基因。通过慢病毒表达的shRNA敲低METTL14或YTHDC1,他们观察到人类ARCOs中摄食相关神经元的产生减少。这表明m6A信号在调节摄食相关神经元生成方面的作用在小鼠和人类之间是保守的。
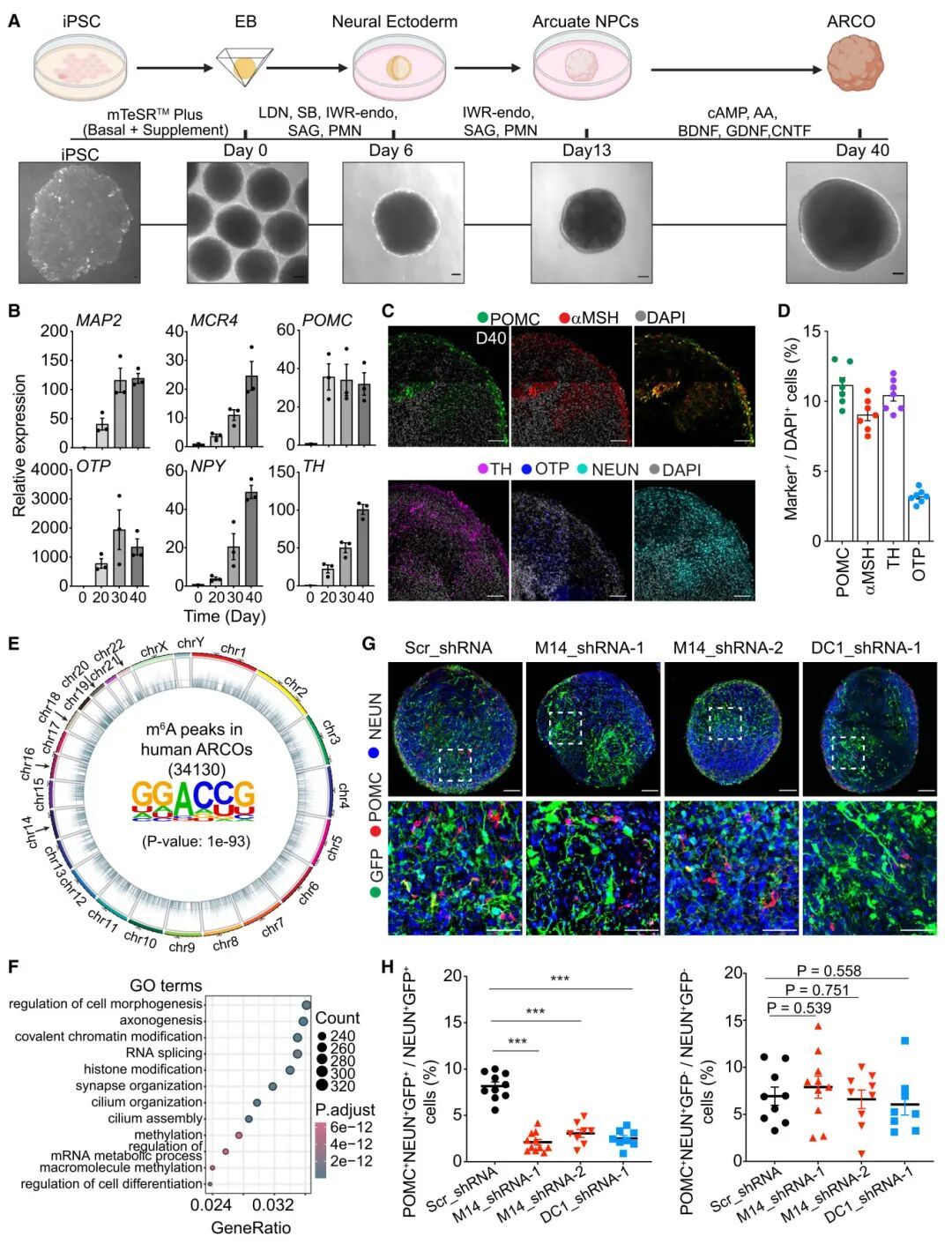
图7 m6A信号调节人类ARCOs中与进食相关神经元的生成
这项研究揭示了m6A修饰在下丘脑神经发生和能量平衡调节中的关键作用。通过多种遗传小鼠模型和人类类器官研究,科学家们确定了Mettl14/Ythdc1依赖的m6A信号通路在小鼠和人类弓状核中对摄食相关神经元的神经发生具有保守作用。这些发现为理解食物摄入和能量稳态的表观转录调节的发育基础提供了重要见解。建立了下丘脑发育缺陷与成年代谢疾病之间的直接联系,揭示了m6A修饰可通过调控特定神经元亚型的发生来影响成年期的体重和脂肪分布。这为肥胖等代谢疾病的发育起源提供了新的理解角度,也为针对RNA修饰的潜在治疗策略开辟了可能性。此外,研究中建立的无饲养细胞人类ARC类器官模型为研究人类下丘脑发育和疾病提供了宝贵工具,使研究人员能够在人类背景下研究RNA修饰和其他表观遗传调控机制。这种模型系统有望促进对人类特异性下丘脑发育过程的理解,并加速针对代谢疾病的个性化医疗方法的发展。
常见细胞污染类型如何辨别及预防解决方法:细胞培养中常见的生物污染类型有7种,分别是细菌污染,支原体污染,原虫污染,黑胶虫污染,真菌污染,病毒污染以及非细胞污染,真菌污染来源,一般是来自实验服,并且具有气候性,多雨······
细胞聚团的原因分析及如何避免:培养物中细胞可能聚集的一些原因包括:1.过度消化、2.环境压力、3.组织分解、4.过度生长、5.污染等;如何避免聚团细胞的生成;首先确认当前细胞生长密度及状态,80%左右的生长密度即可进行······
细胞有空泡原因分析及解决方法:出现细胞空泡情况有1.细胞老化2.培养液错误配制;3.细胞消化时操作不当;4.污染等等,如细胞老化,解决方法,原代细胞使用较低代次进行实验,传代细胞避免传代次数过高···
细胞半换液和全换液操作步骤:第一种方式:细胞全换液;如果是贴壁细胞,可以用全量换液法,直接吸去全部旧培养基,补充足量新鲜完全培养基;第二种方式:细胞半换液;"细胞半换液"又称"细胞半量换液",即弃掉一半旧的培养基,再······
细胞生长缓慢的可能原因有哪些:细胞培养外部因素包括细胞培养基的配方和质量问题,培养条件不理想,污染问题,细胞自身因素包含细胞的健康状态,细胞密度过高或过低,细胞老化现象,细胞特性,当细胞生长出现缓慢的问题时,我······
常用胰腺癌细胞株动物模型及胰腺癌细胞株有哪些:胰腺癌研究中常用的动物模型主要包括化学物质诱导胰腺癌动物模型,基因工程胰腺癌小鼠模型和胰腺癌移植模型,常用的胰腺细胞株MIA-PACA-2人胰腺癌细胞,Capan-2人胰腺癌细······
产品规格:1*10^6
¥3000
产品规格:1*10^6
¥3000
产品规格:1*10^6
¥3000
产品规格:1*10^6
¥3000
上一篇:胃部类器官模型揭示幽门螺杆菌感染中的细胞类型特异性宿主-病原互作
厦门爱恪信生物科技有限公司
手机:15859239971
邮箱:2205839769@qq.com
地址:厦门翔安火炬高新区翔星路96号建业楼D座602

微信公众号
ATCC细胞培养

技术支持
15859239971
Copyright©厦门爱恪信 闽ICP备19027235号-7
公安备案: XML地图
XML地图




 客服QQ
客服QQ